Andy Gray, Bob Goodman, José M. Terán and Barbara Mintzes | 2010 | Download PDF
Original citation: Gray, A., Goodman, B., Teran, J. M., & Mintzes, B. 2010, ‘Using Unbiased Prescribing Information’, in Mintzes, B., Mangin, D., & Hayes, L. (eds.) Understanding and Responding to Pharmaceutical Promotion: A practical guide, Health Action International, Amsterdam; pp 145-161.
The previous chapters discuss a range of promotional techniques, as well as some ways to avoid undue influence of the promotion of medicines on professional practice. A key strategy is to rely only on independent and unbiased information sources as a basis for prescribing and dispensing decisions. Secondly, it is important to know how to judge the strength of this evidence and its applicability: is the design of a study strong enough to support a claimed effect? Is it relevant to your patients? Is adequate information provided on harmful as well as beneficial treatment effects?
This chapter presents basic principles of critical appraisal of clinical trials and concludes with a list of independent information sources as well as criteria you can use to choose information providers. As a busy health professional, you will not always have time to read original studies in order to decide whether a medicine will or will not be useful for your patients. It is also important to be familiar with high-quality, unbiased sources of brief, summarised reviews of the research evidence.
Aims of this chapter
After reading this chapter you should be familiar with:
- The five steps in evidence-based medicine;
- Principles of critical appraisal of studies assessing medicinal treatments;
- Key criteria by which to judge research study quality;
- Sources of reliable, unbiased information on medicines.
Evidence-based medicine and prescribing decisions
Evidence-based medicine aims to base diagnostic and treatment decisions on the full body of existing scientific evidence. Sackett and colleagues (1996) define it as: “…the conscientious, explicit, and judicious use of current best evidence in making decisions about the care of individual patients.” They stress the need to integrate individual, clinical expertise with the best available external, clinical evidence. Practising evidence-based medicine involves five steps:
- Converting the need for information (about prevention, diagnosis, prognosis, therapy or causation) into a clearly stated, answerable question.
- Finding the best evidence with which to answer that question.
- Critically appraising that evidence for its validity (truthfulness), impact (size of the effect), and applicability (usefulness in the practitioner’s clinical practice).
- Integrating the critical appraisal with clinical expertise and with the patient’s unique biology, values and circumstances.
- Evaluating the effectiveness and efficiency of the first four steps and continually improving them.
One of the foundations of evidence-based medicine is the systematic review, which aims to combine all of the existing scientific knowledge about the effects of a specific medical intervention or pharmacotherapy, in order to answer a precisely defined research question. Often a systematic review will also include meta-analysis, a statistical technique that is used to pool research results from included individual trials in order to obtain a quantitative estimate of a treatment’s effects that reflects all of the existing research evidence. Sometimes meta-analysis is not possible because the existing trials differ too much in methods, patient population or evaluated outcomes. When meta-analysis is possible, it provides a powerful tool to obtain a more precise estimate of a treatment’s effects than an individual trial can show, including less common effects and effects in different subgroups of patients. As with any other study design, consideration must be given to actual and potential conflicts of interest that may affect the integrity of the analysis.
Like individual clinical trials, systematic reviews and meta-analyses vary in quality. Some key studies may have been left out, for example. Trials of such poor quality that the results are unlikely to be valid are included nevertheless. Sometimes a systematic review fails to include harmful effects of medicines, leading to a biased view of treatment benefits. Additionally, if negative trials remain unpublished and inaccessible to reviewers, publication bias can lead to inaccurate systematic reviews.
One of the best sources of up-to-date, high-quality, systematic reviews and meta-analyses is the Cochrane Library of Systematic Reviews, produced by the Cochrane Collaboration, a global, non-profit network of researchers who evaluate health-care interventions, including pharmacotherapy. The Cochrane Collaboration has developed a standardised set of methods for systematic reviews and training for reviewers, as well as actively testing, discussing and revising methods as the science of systematic reviews improves.
Box 1: Educational material on evidence-based medicine
| A number of websites provide evidence-based medicine toolkits and tutorials, including: • The Centre for Evidence-Based Medicine (Oxford) http://www.cebm.net/index.asp • The Centre for Evidence-Based Medicine (Toronto) http://www.cebm.utoronto.ca/ • The Evidence-Based Medicine Toolkit http://www.med.ualberta.ca/ebm/ebm.htm |
Limits to the available evidence
| “Evidence-based medicine is valuable to the extent that the evidence base is complete and unbiased. Selective publication of clinical trials — and the outcomes within those trials — can lead to unrealistic estimates of drug effectiveness and alter the apparent risk–benefit ratio.” (Turner E et al, 2008) |
Well-conducted systematic reviews are an important information source because they combine all of the available clinical trial information that addresses a specific question. However, sometimes the type of evidence needed is not available.
There is often a large gap between the situation faced by an individual patient and the body of available research evidence. For example, studies may have been carried out for too short a time. A systematic review of trials of stimulant medicines for attention deficit disorders in children reported an average treatment duration of three weeks (Shachter et al., 2001). Long-term effectiveness remains largely unknown. One 14-month study failed to show an advantage for drug treatment over behavioural therapy or usual care, based on blinded classroom observations (MTA, 1999). However, many children are prescribed these medicines for several years.
Sometimes clinical trial participants are very different – usually younger and healthier – than those commonly prescribed a medicine. The patient in front of you may look more like people who were systematically excluded from clinical trials of a medicine’s effects than trial participants. For example, often elderly people and those with serious health problems are systematically excluded. There may only be placebo-controlled trials with no comparative trials against other active treatments available, making it impossible to know whether a new medicine is better – or worse – than standard treatment. If the medicine has been compared with other treatments, non-equivalent doses may have been used.
Finally, the available published studies may represent only a subset of the scientific evidence about a specific medicine’s effects. For example, Turner and colleagues (2008) found that whereas nearly all published studies (94%) reported that antidepressants were more effective than placebos in treating depression, the picture was very different if all trials, both published and unpublished, were examined. In this case, only 51% – just over half – found the medicines to be better than placebo. This difference reflects both a serious publication bias and reporting bias. Trials in which the medicines looked worse tended not to be published. If a trial was published, the results were often published in a way that made them look more positive than they were.
A second analysis looked at all studies comparing statins (medicines used to lower cholesterol) to one another or to other cholesterol-lowering therapies (Bero et al., 2007). In the 95 industry-funded trials identified, which company sponsored the trial was a strong predictor of which product was found to be superior.
In summary, although the aim of evidence-based medicine is to base treatment decisions on scientific evidence, there are many shortcomings in the scientific evidence, its public availability and its applicability to the situation faced by individual patients.
It is important to keep in mind how strong or weak a body of evidence is, whether there are important gaps in knowledge, and whether the available studies are relevant to your patient. A consistent bias in published clinical trial evidence is that often less evidence is available on harmful than beneficial effects of medicines (Papanikolaou, PN, Ioannidis JP, 2004). This is, in part, simply because not enough people have been included in clinical trials to test whether a rare, serious, harmful effect occurs more often on the medicine than on placebo or comparative treatments. In this case, ‘absence of evidence of harm’ does not mean the same thing as ‘evidence of no harm’.
The truth, the half-truth and nothing like the truth
Promotional materials are not limited to paid advertisements in journals or glossy materials provided by sales representatives. Richard Smith (2005), former editor of the British Medical Journal, has gone so far as to call medical journals part of the marketing apparatus used by the pharmaceutical industry. He is critical of journals’ role in publishing and disseminating biased trial reports that help to stimulate sales. He suggests that “journals should critique trials, not publish them”. Box 2 lists methods companies use to get favourable trial results that are then reported in medical journals.
Box 2. How to get the results you want from a clinical trial
| • Compare your medicine to an inferior treatment; • Compare your medicine to too low a dose of a competitor (for more effectiveness); • Compare your medicine to too high a dose of a competitor (for fewer side effects); • Use too small a sample to show differences; • Use multiple endpoints and publish only those that look best; • Do multicentre trials and publish results only from centres with the best results; • Conduct subgroup analyses and publish only those that are favourable; • Present results that are most likely to impress—for example, reduction in relative rather than absolute risk. (Adapted from Smith R, 2005) |
At the market launch of the Cox-2 inhibitors rofecoxib (Vioxx) and celecoxib (Celebrex), there were high hopes that these medicines would prove safer than other arthritis medicines because of a lower risk of serious gastrointestinal bleeding. The first trials testing this hypothesis were published after both medicines had begun to achieve ‘blockbuster’ sales, based, in large part, on a promise of greater safety.
What happened when study results proved differently? The published clinical trial reports of both the Vioxx Gastrointestinal Outcomes Research (VIGOR) trial (Bombardier et al., 2000) and the Celecoxib Long-Term Arthritis Study (CLASS) trial (Silverstein et al., 2000) claimed safety advantages. In both cases, this was based on incomplete reporting of clinical trial data.
The VIGOR trial aimed to assess rates of serious gastrointestinal bleeding and found a lower rate with rofecoxib than naproxen. However, more patients on rofecoxib experienced serious cardiovascular events. The authors argued that this difference was due to the cardio-protective effect of naproxen and the published report mainly discussed gastrointestinal bleeding, although more patients were affected by the increased cardiovascular risks.
In late 2004, rofecoxib was withdrawn globally because of increased risks of heart attack and stroke. Soon after, the journal editors published an ‘expression of concern’ (Curfman et al., 2005) because three heart attacks among rofecoxib users were not included in the VIGOR report or the manuscripts they had reviewed. The study’s academic authors argued in a rebuttal that they had acted correctly because of a pre-specified study protocol. Curfman and colleagues point out the larger picture: “Because these data were not included in the published article, conclusions regarding the safety of rofecoxib were misleading.” (Curfman et al., 2006).
In a similar case, data published on the CLASS study (Silverstein et al., 2000) differed from the data presented to the US Food and Drug Administration (FDA) (Hrachovec and Mora, 2001; Wright et al., 2001). Trial results were reported at six months and did not refer to the full, longer study period. The first six months looked better for celecoxib. However, when the full 12-month trial data were examined, most ulcer complications had occurred in the second six months, meaning that there was no significant safety advantage (Jüni, 2002). Again, the authors defended their work, but did acknowledge that “we could have avoided confusion by explaining to the JAMA editors why we chose to inform them only of the 6-month analyses, and not the long-term data that were available to us when we submitted the manuscript.” (Silverstein et al., 2001).
Interpreting the numbers
In Box 2, Smith mentions the use of relative rather than absolute risk differences as a common misleading presentation of data. When only relative risks are reported, a small difference in a rare event can be made to look clinically significant. The following example illustrates this practice and also the way in which an alternative metric, the number needed to treat (NNT) can be calculated.
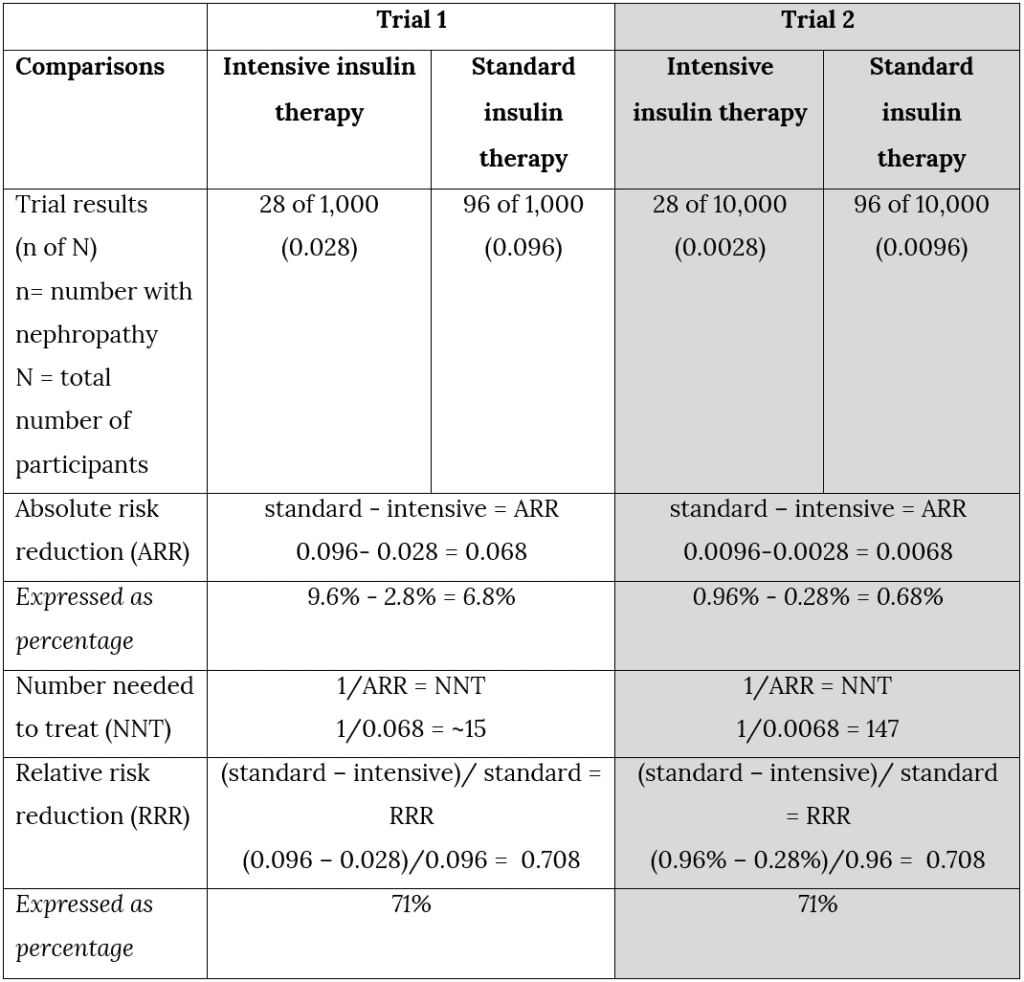
Table 1 compares the results of two imaginary trials (‘Trial 1’ and ‘Trial 2’) on the effects of intensive versus standard insulin treatment on diabetic nephropathy. It illustrates how different relative versus absolute risk reductions can look.
Here are the results of Trial 1:
- 28 of 1,000 patients on intensive insulin developed diabetic nephropathy;
- 96 of 1,000 patients on standard insulin developed diabetic nephropathy.
The absolute risk reduction is calculated by subtracting the event rate in the experimental group from the event rate in the control group. As Table 1 illustrates, in Trial 1, the absolute risk reduction is 9.6% – 2.8% = 6.8%. In a two-year trial, it would mean that around 7 fewer patients on intensive therapy out of each 100 patients treated would experience nephropathy over a two-year period.
Imagine that only one-tenth as many people developed diabetic nephropathy over a two-year period. This is the situation in Trial 2, (the right-hand column):
- 28 of 10,000 patients on intensive insulin developed diabetic nephropathy;
- 96 of 10,000 patients on standard insulin developed diabetic nephropathy.
The absolute risk reduction is 0.96% – 0.28% = 0.68%. This is much less impressive, and a physician would probably be much less likely to recommend intensive treatment. Out of every 100 people treated, one fewer patient would develop diabetic nephropathy each two years.
Number needed to treat (NNT) is another way of expressing the same results and using the concept of absolute risk. The focus is on the individual patient’s probability of benefit. The NNT shows the number of patients with diabetes who need to be treated with the intensive regimen in order to avoid 1 additional case of diabetic nephropathy. It is calculated by taking the reciprocal of the absolute risk reduction:
For Trial 1: NNT = 1 ÷ 0.068, or ~15.
In other words, 15 patients need to be treated for 1 to benefit. For Trial 2: 147 patients must be treated for 1 to benefit.
If relative risk reduction is used instead, these differences disappear and the results look far more impressive for both trials. Relative risk reduction is a measure of the difference between the two rates ‘relative to’ the rate for standard treatment:
Trial 1 (9.6-2.8) ÷ 9.6 = 71%
Trial 2 (0.96-0.28) ÷ 0.96 = 71%
It is not hard to guess why this measure is often used to advertise benefits: “reduce your patients’ risk of diabetic nephropathy by 71%”. This is accurate – both for Trial 1 and Trial 2 – but without mentioning the absolute differences it can also be highly misleading. Table 1 summarises the measures for absolute risk reduction, numbers needed to treat, and relative risk reduction for these two imaginary trials.
Table 1: Why report absolute risk reductions? An illustration
For a physician or pharmacist making treatment decisions it is always important to know not just the relative differences between two approaches to treatment, but also absolute differences and how likely a patient is to benefit. When this probability is very small sometimes medicine treatment may be as likely or almost as likely to lead to harm. Allenso-Coello and colleagues (2008) use the example of the hormonal medicine raloxifene for prevention of osteoporosis. An advertisement claimed a 75% relative risk reduction for vertebral fractures (see Chapter 3). In the population group targeted, fracture rates are expected to be less than 1% per year. Allenso-Coello et al. calculate the number needed to treat as 133 (95% confidence interval 104 to 270) for 3 years to prevent one fracture. Raloxifene also leads to an increase in thromboembolic events (deep-vein thrombosis and pulmonary embolism). With an absolute risk increase of 0.7%, the number needed to harm is 143 for 3 years. The number needed to harm is the number of patients that would have to receive this medicine in order for one additional person to develop the adverse effect of interest. An independent bulletin points out that the magnitude of benefit is similar to the magnitude of harm (Therapeutics Initiative, 2000).
If the benefits of medicines are reported only as relative risk reductions and harmful effects only as absolute risk increases, it is very hard to compare the effects directly, or to know that sometimes – as in the case above – the likelihood of benefit and harm are very similar.
Critical appraisal of studies of medicinal treatments
The term ‘critical appraisal’ refers to methods used to evaluate the strength of a study’s design, the way the study was carried out, and the reporting of results, in order to judge the results’ validity. One rule for critical appraisal is to pay close attention to the methods section of a clinical trial report and not too much attention to the study abstract, which is often misleading. Unfortunately, busy clinicians often do exactly the opposite. Critical appraisal of a clinical trial’s results, and its relevance to your patients, starts with a few key elements:
- Type of studies: evidence of benefit must be based on the strongest possible research evidence, generally double-blind, randomised, controlled trials.
- Was selection truly random? Sometimes methods are used that allow some selection of participants for different treatment groups.
- Were some patients screened out in a ‘run in’ period (for example, those who responded less well to treatment or better to placebo); this creates a biased subset of trial participants.
- Were patients, clinicians and assessors adequately ‘blinded’ to treatment allocation? Methods should be described in detail and ideally the adequacy of blinding tested by asking patients and clinicians to guess what treatment they are on.
- Is the sample size adequate? The sample size calculation should be described in the trial.
- Type of participants: these should be similar to the types of patients encountered in normal clinical care. For example, if a medicine is often used by the elderly and they were excluded in a trial, this is a problem, or if patients in a trial are much healthier (for example, if patients with co-morbidities were unnecessarily excluded) or much more ill than those encountered in clinical care.
- Types of comparisons: the study should compare a new medicine with standard treatment for the same condition or with a placebo, only if there is no standard treatment. If a medicine is in an existing class with a specific mechanism of action, it should be compared with other medicines in the class. The dose should also be comparable.
- Accounting for all patients in the trial: analysis should be by ‘intention-to-treat’, and include all patients randomised to each treatment arm, regardless of whether they discontinued early or not.
- Type of outcome measures: the main focus should be on health effects of importance to patients’ lives. Serious morbidity and mortality are given priority over symptom-free, physiological effects.
- Funding source and conflicts of interest: if the study was funded by a pharmaceutical company, were any procedures in place to prevent sponsor involvement in study design, analysis of data and reporting?
Box 3: Useful websites on evidence-based medicine and critical appraisal skills
| Centre for Evidence-Based Medicine (CEBM), Oxford, UK http://www.cebm.net/index.aspx?o=1011 CEBM tools and worksheets for critical appraisal http://www.cebm.net/index.aspx?o=1157 The ‘evidence-based toolkit’ from the University of Alberta, Canada http://www.ebm.med.ualberta.ca/ Similar resources from the University of Toronto http://www.cebm.utoronto.ca/ Organisations that carry out systematic reviews of clinical evidence National Institute for Clinical Excellence (NICE), UK http://www.nice.org.uk/ Scottish Intercollegiate Guidelines Network http://www.sign.ac.uk/ US Agency for Healthcare Research and Quality (AHRQ) http://www.ahrq.gov/ and the AHRQ web portal for clinical guidelines http://www.guideline.gov/ The Cochrane Collaboration – an international organisation that carries out systematic reviews of health care interventions (includes database of systematic reviews) (Source: http://www.cochrane.org/) |
The use and abuse of statistics in advertising
It is useful to know some of the
common tricks that may be used in advertising and promotion to misrepresent the
scientific evidence. Someone glancing at an advertisement or other promotional
materials may not immediately notice them, but once alerted, they become much
more obvious. Box 5 is an overview of some of the more common techniques used,
produced by a regional drug and therapeutics committee in the UK.
Box 4: The Hitch-hikers’ guide to promotional drug literature
Beauty is only skin deep – be wary of skilful and seductive graphics designed to grab your attention and distract from the actual content. Look out for irrelevant photographs, e.g. advertisers are more likely to use glamorous, well-dressed women to sell oral contraceptives, and harassed mothers to sell antidepressants.
What’s the point – strip away the multicoloured hype and ask yourself: what does the advert really say? Does it actually say anything? Is it merely a gimmick to reinforce a brand name? Is the drug really new, or the consequence of “molecular roulette”?
Examine the claims – check the original evidence on which the claim is based. If possible, compare statements, quotations and conclusions with the original article. Direct misquotation is not unknown, nor is quoting out of context, nor citing of studies with inadequate methodology.
The great picture show – these are the favourite tools of the advertiser – points to check: (i) make sure the axes start at zero. Axial distortion may make an insignificant difference “look significant”; (ii) Lines of graphs should not be extended beyond plotted points, and that there is an indication of variance (e.g. standard error bars); (iii) “Amputated” bar charts (similar effect to (ii)); (iv) Logarithmic as opposed to numerical plots.
“Lies, damned lies and statistics” – always be suspicious of statistics. Most readers have only a very basic knowledge of statistics. Beware of BIG percentages from small samples. “p” values are only worthwhile if data have been properly and accurately collected in a well-designed trial and the correct statistical test used for analysis.
Non sequiturs – this involves positioning two irrelevant statements in a manner implying a relationship – commonly, pharmacokinetic data from single dose studies in young healthy volunteers and the implication that this will apply to elderly patients with multiple pathology in a chronic dosage schedule.
References – if there are any – examine the list of references carefully. Be suspicious if references are old or from obscure or unfamiliar foreign journals (some journals exist only to publish drug company sponsored papers). “Data on file”, “Symposium proceedings”, “To be published” or “Personal communication” should also ring warning bells. Remember an isolated quote taken out of context can alter the real conclusion of the reference.
A scanned image of the original version published in the Essential Drugs Monitor No. 17, 1994, can be accessed at: http://www.healthyskepticism.org/library/EDM/17/Page24.pdf (West Lothian Drug and Therapeutics Committee, 1994)
Conclusion
This chapter has provided a brief introduction to evidence-based medicine, including some of the difficulties involved in interpreting clinical trial reports and techniques that can be used to misrepresent or to more accurately represent results. We would also like to conclude with lists of resources and a positive, alternative approach to the most common treatment decisions you will encounter.
Develop a personal formulary
Nobody has time to look up all the evidence on all available treatments each time a patient comes in for care or to search the Cochrane Library or another information source for the most relevant systematic reviews. In reality, doctors see many patients for many of the same health problems, day after day. Although there are differences between patients both in general health and treatment preferences, it is possible to develop a list of medicines and other treatments that can be helpful to most patients with a specific condition, most of the time. In primary care, around 50 to 100 medicines can meet nearly all of patients’ health needs. It is especially useful to develop a personal list of medicines to treat the most commonly encountered health problems in most patients.
Box 5: Five key steps required for rational prescribing decisions
- Define the patient’s problem.
- Specify your treatment objective (i.e. what you are trying to achieve, in how long?)
- Make an inventory of possible treatments. This can include drug and non-drug options, information and advice, watchful waiting and the option not to treat.
- Choose your P-treatment (personal treatment) based on efficacy, safety suitability and cost.
- Verify that the ‘P-treatment’ is appropriate for this patient.
(de Vries et al., 1994)
Resources
Some sources of independent information on medicines
Below are a few examples of English-language, non-promotional information on medicines. Sources will, of course, vary depending on where one is practising (as approved medicines and regulatory agencies will also vary), so it is not the intention here to review and compare the world’s sources of unbiased pharmaceutical information, only to give a very small sampling. Note that none of these sources comes with a free lunch.
International Society of Drug Bulletins
Founded in 1986, ISDB is a network of drug bulletins and journals whose members are “financially and intellectually independent of the pharmaceutical industry.” The aim of ISDB is to assist in the development of drug bulletins and facilitate cooperation of bulletins in different countries. For more information, visit: http://www.isdbweb.org
The Medical Letter
Published in the US since 1959, it is one of the better known bulletins in this country. Independent of the pharmaceutical industry, it gives practical, concise recommendations, accompanied by information on cost, adverse effects and comparisons with other medicines. In addition to a paper version, it is available online and for PDA. It is available by subscription, see http://medletter.com
Prescrire International
This is the English-language version of the French drug bulletin La revue Prescrire. Prescrire provides independent information on new medicines and indications, adverse effects, cost comparisons, as well as treatment guidelines. It is available by subscription and is an ISDB member, see http://www.prescrire.org
Drug and Therapeutics Bulletin
Started in 1963, this is a monthly UK publication giving independent evaluations of, and practical advice on, individual treatments and the management of disease. It is available by subscription, see: http://www.dtb.org.uk
Prescriber’s Letter
This independent monthly newsletter is published in the US. It is available as a printed newsletter, by subscription, with online and PDA versions, see: http://www.prescribersletter.com
Therapeutics Letter
This publication was established in 1994 by the Department of Pharmacology and Therapeutics at Canada’s University of British Columbia “to provide physicians and pharmacists with up-to-date, evidence-based, practical information on rational drug therapy.” It is available free of charge. The letter is an ISDB member, see: http://www.ti.ubc.ca/
Martindale Complete Drug Reference
First published in 1883, this reference book covers medicines, veterinary and investigational agents, herbal medicines, as well as toxic substances. It is available in online and PDA versions (although it is not free), see: http://www.medicinescomplete.com/mc/
Worst Pills Best Pills
Produced by the Public Citizen Health
Research Group, this is the only US bulletin that is a member of ISDB. It is
primarily intended for patients. The newsletter is available online and on
paper, by subscription, see: http://worstpills.org
Guides to critical appraisal of the research evidence
Montori VM et al. (2004).Users’ guide to detecting misleading claims in clinical research reports. British Medical Journal 329:1093–1096.
Guyatt G et al. (1998). Interpreting treatment effects in randomised trials. British Medical Journal, 316:690–693.
Greenhalgh T (1996). Is my practice evidence-based? British Medical Journal, 313(7063):957-958.
Greenhalgh T (1997). How to read a paper. The Medline database. British Medical Journal, 315(7101):180-183.
Greenhalgh T (1997). How to read a paper. Getting your bearings (deciding what the paper is about). British Medical Journal, 315(7102):243-246.
Greenhalgh T (1997). Assessing the methodological quality of published papers. British Medical Journal, 315(7103):305-308.
Greenhalgh T (1997). How to read a paper. Statistics for the non-statistician. I: Different types of data need different statistical tests. British Medical Journal 315(7104):364-366.
Greenhalgh T (1997). How to read a paper. Statistics for the non-statistician. II: “Significant” relations and their pitfalls. British Medical Journal 315(7105):422-425.
Greenhalgh T, Taylor R (1997). Papers that go beyond numbers (qualitative research). British Medical Journal, 315(7110):740-743.
Greenhalgh T (1997). How to read a paper. Papers that tell you what things cost (economic analyses). British Medical Journal, 315(7108):596-599.
Greenhalgh T (1997). How to read a paper. Papers that report drug trials. British Medical Journal, 315(7106):480-483.
Greenhalgh T (1997). Papers that summarise other papers (systematic reviews and meta-analyses). British Medical Journal, 315(7109):672-675.
Greenhalgh T (1997). How to read a paper. Papers that report
diagnostic or screening tests. British
Medical Journal 315(7107):540-543.
Student exercises
- Assessing advertisements
Select an advertisement from a medical journal or news magazine, and as a group, critically examine the advertisement by using the elements listed in “The Hitch-hiker’s Guide to Promotional Drug Literature” (see Box 5). In particular:
- Check if any
statistics are quoted or graphically represented. Were these done
appropriately?
- Check if the results are represented as a relative risk reduction or an absolute risk reduction – if an RRR was presented, can the ARR and NNT be calculated?
- Check if the research cited is retrievable from your library. Was it quoted as “data on file”? If so, what does this mean?
Share your results with the class and discuss how this would affect your view of the medicine being advertised.
References
Alonso-Coello P, Garcia-Franco AL, Guaytt G et al. (2008). Drugs for pre-osteoporosis: prevention or disease-mongering? British Medical Journal 2008; 336: 126-129.
Bero L. Oostvogel F. Bacchetti P. Lee K. Factors associated with findings of published trials of drug-drug comparisons: why some statins appear more efficacious than others. PLoS Medicine / Public Library of Science 2007; 4(6):e184.
Bombardier C, Laine L, Reicin A et al. (2000). Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis. New England Journal of Medicine 343:1520-8.
Curfman GD, Morrissey S, Drazen JM (2005). Expression of concern: Bombardier et al., Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis. New England Journal of Medicine 353:2813-4.
Curfman GD,
Morrissey S, Drazen JM (2006). Expression of concern reaffirmed.
New England Journal of Medicine 354(11):1193.
De Vries TPGM, Henning RH, Hogerzeil HV et al. (1994). Guide to good prescribing: A practical manual. Geneva, World Health Organization. WHO/DAP/94.11.
Hrachovec JB, Mora M (2001). Reporting of 6-month vs 12-month data in a clinical trial of celecoxib. Journal of the American Medical Association 286(19):2398.
Jüni P (2002). Are selective COX 2 inhibitors superior to traditional non steroidal anti-inflammatory drugs? British Medical Journal 29;324:1287-8.
The MTA Cooperative Group (1999). A 14-month randomised clinical trial of treatment strategies for attention-deficit/hyperactivity disorder. Archives of General Psychiatry;56:1073-1086.
Papanikolaou PN, Ioannidis JP (2004). Availability of large-scale evidence on specific harms from systematic reviews of randomized trials. American Journal of Medicine 117(8):582-9, 15 Oct.
Sackett DL, Rosenberg, WMC, Gray JAM et al. (1996). Evidence based medicine: what it is and what it isn’t. British Medical Journal 312(7023):71-72.
Schachter HM, Pham B, King J, et al. (2001). How efficacious and safe is short-acting methylphenidate for the treatment of attention-deficit disorder in children and adolescents? A meta-analysis. Canadian Medical Association Journal.;165:1475-1488.
Silverstein FE, Faich G, Goldstein JL et al. (2000). Gastrointestinal toxicity with celecoxib vs nonsteroidal anti-inflammatory drugs for osteoarthritis and rheumatoid arthritis: the CLASS study. A randomized controlled trial. Celecoxib Long-term Arthritis Safety Study. Journal of the American Medical Association, 284(10):1247-55.
Silverstein FE, Simon L, Faich G (2001). Reporting of 6-month vs 12-month data in a clinical trial of Celecoxib – in reply. Journal of the American Medical Association, 286(19):2399-2400.
Smith R (2005). Medical journals are an extension of the marketing arm of pharmaceutical companies. Public Library of Science Medicine 2(5):e138.
Therapeutics Initiative (2000). New drugs V. Orlistat (Xenical), Raloxifene (Evista), Spironolactone (Aldactone). Therapeutics Letter, Issue 34, March-April, ( http://www.ti.ubc.ca/en/TherapeuticsLetters, accessed 29 April 2009).
Turner E et al. (2008). Selective publication of antidepressant trials and its influence on apparent efficacy. New England Journal of Medicine, 2008; 358:252-260.
Wright JM, Perry TL, Bassett KL et al. (2001). Reporting of 6-month vs 12-month data in a clinical trial of celecoxib. Journal of the American Medical Association, 286(19):2398-400.